Click now to listen to RCA’s Director of Regulatory Affairs, Jordan Elder, in this RCA Radio audio update:
The European Parliament recently voted for a timeline extension to MDR regulation, including an extended timeline for medical device regulatory submission. The 537-3 vote by members of the official body provides a final approval to extend MDR deadlines for compliance. Included in the legislation is revisions to regulatory submission rules for certifying medical devices. Additionally, the timeline includes new dates for both Regulation (EU) 2017/745 (MDR) and Regulation (EU) 2017/746 (IVDR).
Regulated Companies
European officials extended the MDR timeline to include legacy devices with existing certificates under the MDD to stay on the market until 2024. Industry executives have suspected for some time that the EU medical device backlog of submissions would lead to this type of scenario. Equally important, many EU health ministers voiced their opinion during a December 2022 session medical device shortages would occur without a MDR timeline extension.
MDR Timeline
Manufacturers now have until 2027 or 2028 to have medical devices certified and approved under MDR guidelines. The timeline includes new dates for both high-risk medical device products and low risk medical devices. For medical devices covered by a certificate or a declaration of conformity issued before 26 May 2021, the transition period to the new rules is extended from 26 May 2024 to:
- 31 December 2027 for Class III and IIb;
- 31 December 2028 for Class IIa and I.
- 26 May 2026 for Class III implantable custom-made devices
The original “sell-off’ date requirement that was required under the MDR has been removed
Need help with your MDR or IVDR transition? Talk to our Experts →
IVDR Timeline
Meanwhile, the EU Commission has previously recommended the IVDR application date be extended due to the EU notified body bottleneck.
- General EU MDR Class 1 Low-risk devices that are non-measuring, non-sterile, non-reusable, non-surgical, and that do not require review from a notified body will still go into effect in 2022.
- Non-sterile Class A and B Devices (low risk) – May 26, 2022
- Class D (Highest Risk) – May 26, 2025
- Class C (Medium Risk) – May 26, 2026
- Sterile Class A and B Devices (low risk) – May 26, 2027
Industry Reaction
Life science media outlets have reported extensively on the change since it was proposed at the meeting.
Stella Kyriakides, the European health commissioner, first proposed postponing the current MDR deadline dates during the EPSCO council meeting in Brussels. The health commissioner projected around 23,000 devices and 1,500 IVDs are currently approved and certified under MDD, but have not yet transitioned to the new MDR regulation. These medical devices are likely to expire in 2024 and 2025,
“The transition to the new rules has been slower than we anticipated,” said Kyriakides. “The pandemic, shortages of raw materials caused by the Russian invasion against Ukraine and low notified body capacity has put a strain on market readiness.”
Medical Device Manufacturers
Additionally, many unique viewpoints have emerged about the current landscape and the impact of recertification.
“If the three-year deadline is truly unattainable, these extensions could prevent devices that are perfectly safe for use from being taken off the market because they were unable to get recertified in time,” said Alexandra Murdoch, a medical analyst at GlobalData.
Murdoch added both medical device manufacturers and suppliers must now deliver MDR regulatory documentation for market approval, including data about materials used in medical device manufacturing.
Medical Device Safety
Both Pinto & Rocha have documented the MDR proposal concerns that “only medical devices considered safe will benefit from the extension”. This includes medical device manufacturers that have begun the process of submission and certification under the MDR.
Further, the Commission has recognized the ongoing need for patient safety and proposed a 2023 pilot project for medical device manufacturers. For example, expert panels to advise manufacturers with qualified scientific advice about devices that help treat rare diseases.
Medical Device Shortage
Ireland’s minister of health, Stephen Donnelly, supported the change based on COVID-19 procurement and preventing future medical device shortage scenarios.
“Participation in the EU COVID-19 vaccine strategy has allowed us to conduct the largest immunization program in our country’s history, saving countless lives and enabling the resumption of normal social and economic life.” said Donnelly.
“This measure needs to be adopted and take effect without delay to ensure that the devices our citizens and health systems rely on remain available.”
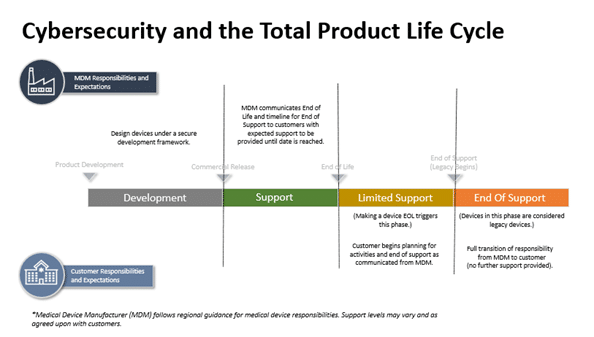
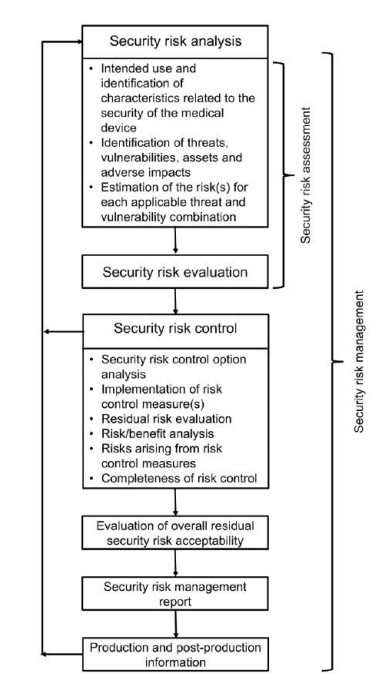
Cybersecurity
By contrast, it is still unknown how different types of EU legislation will regulate connected medical devices. There is concern across the industry about which legislation may take precedence and the level of postmarket surveillance data needed.
The European Commission (EC) published a proposal for a Cyber Resilience Act (“CRA”) to strengthen cybersecurity across medical device interoperability. Both Wright & Wenzel have documented this legislation does not consider MDR to impose as many obligations on medical device manufacturers. Further, the commentary suggests the EC may not require as much documentation about unknown vulnerabilities are not present for medical devices.