















Published Articles
Lisa L. Michels, General Counsel and regulatory affairs expert at Regulatory Compliance Associates® Inc., discusses how the global medical device regulatory landscape is impacted by international law.
The global medical device regulatory landscape is constantly evolving as governing bodies and/or agencies worldwide continue their efforts to harmonize and streamline rules. Over the last several years, many of these regulatory bodies have joined forces to ensure consistency in the interpretation, application, and implementation of relevant medtech regulations and globally recognized consensus standards.
Regulatory Submission
While such efforts have considerably improved the product registration process overseas, harmonization in some cases has made it difficult for some device firms to stay abreast of the planet’s evolving regulatory landscape. To remain competitive in this challenging environment, medtech companies stay ahead of the competition by proactively planning for new and/or changing global regulations. This helps agile organizations successfully launch existing, modified, or novel products in new target markets quickly.
Medical Device Registration
Most companies want their products to be registered in as many global markets as possible, but this lofty goal introduces certain regulatory challenges. From a general business perspective, global product registration in numerous foreign markets obviously makes sense.
Companies possessing the legal authority to sell product(s) in certain regions where competitors are not authorized to do so often distinguishes those organizations as leaders or industry trailblazers. This approach is not always logical or practical, however.
Regulatory Pathway
Product registration should be based on a proactive strategic regulatory analysis. Understanding the available regulatory pathway options can help properly determine whether registration in a particular market. Ultimately, the team is challenges with the regions that make sense for the product and business as a whole.
The most common international medical device registration challenges are primarily based on the following considerations:
- Understanding and applying country-specific medical device regulations across global markets
- Timely implementation of country-specific medical device regulations in target markets
- Costs associated with global product registration(s), which may include (but are not limited to) in-country agent representation, in-country product testing, and in-country collection of clinical data
- Excessive delays and/or long lead times for regulatory reviews needed for global product registration(s) in target markets
Device Classification
These challenges are much easier to manage when they are properly assessed well before starting the product registration processes in certain global markets. Based on the device classification and type of product, there may be more (or less stringent) requirements that must be achieved in specific countries.
The risk level of the product and the existence of similar approved products in a particular market can complicate the product registration process in certain countries.
Medtech Programs
Many medtech companies typically tend to focus their medical device product registration efforts on seven (7) primary global markets: the United States, Canada, European Union (EU), Australia, Brazil, China, and Japan. Due to ongoing harmonization efforts, these countries are working together to streamline the registration processes and improve their consistency.
Depending on the company’s overall product launch strategy, device registration in emerging markets has become common in recent years. In certain cases, product registration may be less challenging in these markets because regulations have not yet been fully developed.
Pre-Clinical Testing
The applicable medical device regulations in target markets each pose their own product registration challenges. Therefore, it is imperative that companies plan for all target market requirements early in the product development process—ideally at the idea stage.
Understanding and preparing for the hurdles associated with pre-clinical testing, clinical testing, performance testing, safety testing, labeling, and compliance with harmonized standards, can help companies overcome possible challenges with the product registration process in each potential target market.
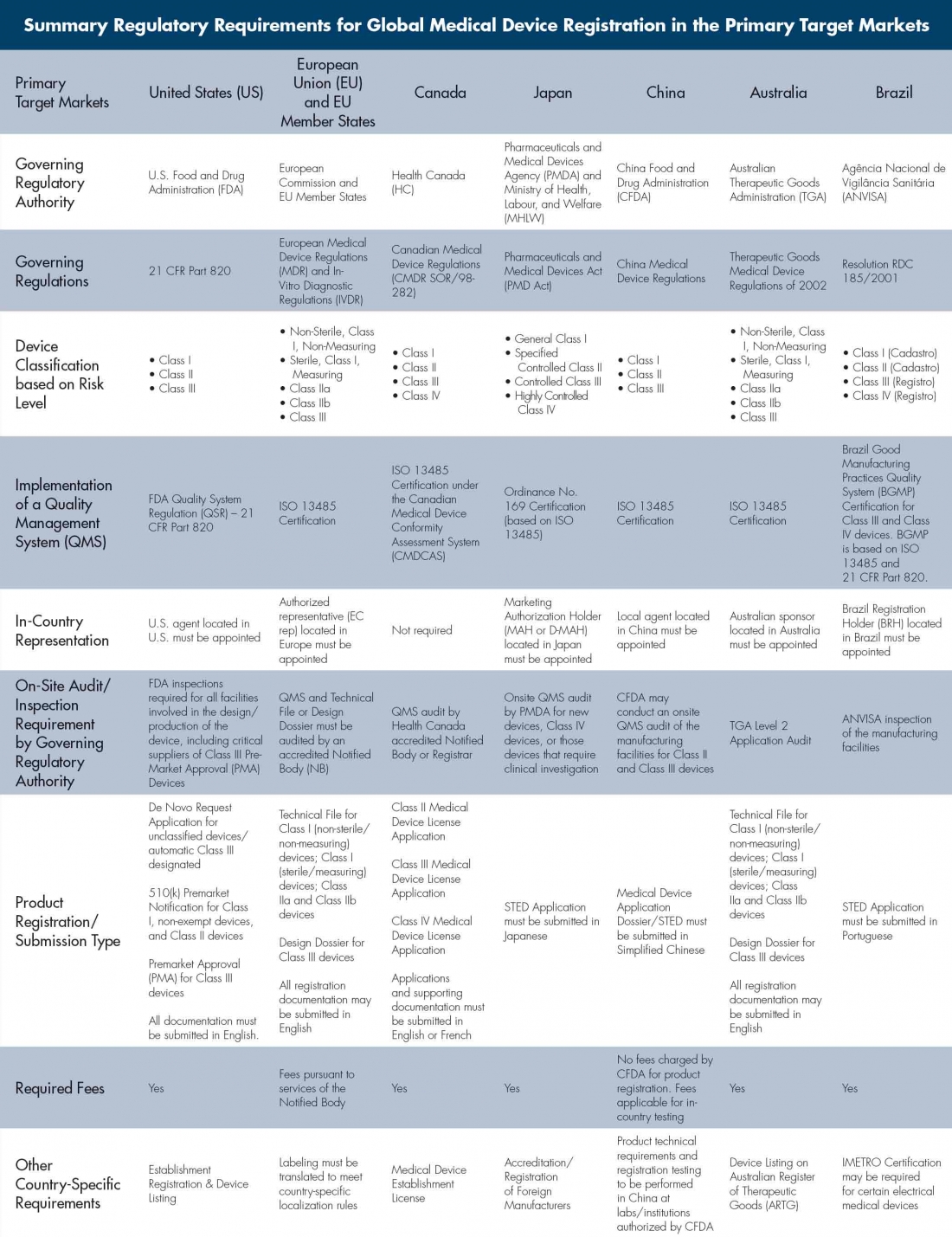
Table: Summary Regulatory Requirements for Global Medical Device Registration in the Primary Target Markets
Regulatory Compliance
Achieving product registration compliance in each target market may seem like a daunting endeavor, but it’s not truly as complicated as it sounds. There are many similarities between the various regulations, as outlined in the table at right. Although the table represents only a subset of requirements, the country-specific mandates applicable to product type in each of the target markets must be carefully reviewed and assessed.
The most effective method to accomplish this task is to prepare a global regulatory strategy for the product(s), which may ultimately be used as a gauge to determine the likelihood of successfully launching a device in a particular market.
Regulatory Strategy
A global regulatory strategy is a documented analysis that defines the overall business objectives and requirements necessary for foreign product registration. A regulatory plan, on the other hand, is a comprehensive report based on the foundational regulatory strategy that outlines all product- and country-specific registration mandates in particular markets.
The regulatory strategy and plan may be separate stand-alone documents or combined into a single global strategy and planning file. Regardless of format, however, their purpose is the same—to proactively document and plan for the implementation of applicable regulatory requirements associated with specific products in each target market.
Regulatory Audit
Keep in mind that these regulatory strategy and planning document(s) are not static. They must be updated and revised accordingly when changes are made either to the product itself or to the device’s launch strategy. A revision would be required, for example, if target markets are added or removed from the launch strategy. Any regulatory body will want to understand the risks made to existing products and regulations (particularly when new rules are introduced that can significantly affect or completely alter product registration mandates).
The format of the regulatory strategy and planning document ultimately depends on the company. Some organizations encourage the use of a specific format or template to document the regulatory strategy and/or plan. Other firms are more liberal about format.
Medical Device Regulatory
Fundamentally though, the setup of the regulatory strategy and planning document is not as important as the goal: To initiate and maintain robust, proactive, regulatory planning efforts as early as possible, and to effectively monitor any changes in the scope to avoid unexpected and costly delays. The key elements of a global regulatory strategy and planning document are also outlined in the table.
Medical Device Global Regulatory Strategy and Planning Document
Detailed Product Summary
- Detailed description of the product including its accessories, components, and software (if applicable)
- Describe the product requirements, including technological/functional/performance/clinical requirements of the product (e.g., what the product does and how it works/mode of operation)
- Indications for use/intended use
- Target population
- Labeling requirements
- All proposed marketing claims and requirements for claim substantiation
- Risk level/product classification/applicable product codes
- Predicate(s) and/or similar products on the market in each target country
Governing Regulatory Body and/or Agency in Each Target Market
- Planned target markets for product launch
- Applicable laws, regulations, standards, and relevant guidance for the product
- Analysis of harmonized requirements and standards for the product across all target markets
- Reimbursement requirements and other country-specific considerations
Proposed Regulatory Pathway and Product Registration Requirements in Each Target Market
- Regulatory submission/product registration requirements, including all applicable product testing requirements (e.g., pre-clinical testing, animal testing, clinical testing, performance testing, safety testing, etc.)
- Timeline to obtain clearance/approval
- Costs associated with global product registration
- Required resources (internal/external)
- Pre-market submission/early interaction, discussion, or consultation with governing regulatory agency or authority regarding product and proposed regulatory pathway
General Requirements for Product Registration in Each Target Market
- Implementation of a Quality Management System (QMS) compliant with applicable regulations and/or standards
- QMS audit/inspection of manufacturing facilities by the regulatory agency
Country-Specific Requirements for Product Registration in Each Target Market
- Appointment of an in-country, local agent/representative/sponsor/distributor to manage the product registration on behalf of a foreign manufacturer
- In-country product testing requirements and product samples
- In-country clinical trial/data collection requirements
- Product registries/databases
- Labeling requirements
- Language translations for labeling [e.g., instructions for use (IFU), operator or user manual, labels, etc.]
Other information as applicable for the specific type of product
Recommendations for Successful Navigation
The key to successfully navigating through the regulatory challenges of global product registration in the international marketplace starts and ends with diligent and proactive planning.
Product registration efforts should focus on markets where companies can leverage device submission documentation for previously approved applications. If, for example, a company has U.S. Food and Drug Administration clearance and/or CE mark approval for a product, it is often easier to register that exact device in another global market that recognizes the same (or similar) harmonized requirements and standards.
Companies should ensure their products are tested and comply with globally recognized consensus standards. A sampling of medical devices standards recognized in many international markets include:
- EN 1041:2008—Information supplied by the manufacturer of medical devices
- EN ISO 13485:2012—Medical devices – Quality management systems – Requirements for regulatory purposes (ISO 13485:2003)
- EN ISO 14155:2011—Clinical investigation of medical devices for human subjects – Good clinical practice (ISO 14155:2011)
- EN ISO 14971:2012—Medical devices – Application of risk management to medical devices (ISO 14971:2007, Corrected version 2007-10-01)
- EN 60601-1:2006—Medical electrical equipment—Part 1: General requirements for basic safety and essential performance
- EN 60601-1-2:2007—Medical electrical equipment—Part 1-2: General requirements for basic safety and essential performance – Collateral standard: Electromagnetic
- EN 60601-1-6:2007—Medical electrical equipment—Part 1-6: General requirements for basic safety and essential performance – Collateral Standard: Usability
- EN 62304:2006—Medical device software – Software life-cycle processes
A comprehensive global regulatory strategy and planning document must be prepared that clearly identifies all county-specific requirements necessary for successful product registration in each target market. In addition, companies should set realistic and attainable goals for timely product launches only in the markets that align with their overall global business strategy. Commercialization will only be successful if the product is a logical fit for the chosen market.
Perhaps most importantly, medtech firms must not register a product in a certain market simply because they can do so. Rather, they should make strategic and informed regulatory decisions about their product launch plans so they can avoid the most common regulatory challenges impacting this highly competitive industry.
About RCA’s Medical Device Consulting Services
The regulatory compliance process surrounding the medical device industry involves a strict adherence to pre/post market information throughout a device’s life-cycle. Even a single compliance issue you have can turn into a significant effect on your business. Regulatory Compliance Associates medical device consultants can help guide you through any stage of this strategic process, with capabilities during product development through the regulatory clearance/approval of your product.
Our team of over 500 medical device consulting Experts — including former FDA officials and regulatory compliance leaders in the field of medical device regulation — will work with your company to create a quality assurance and regulatory compliance approach tailored to your products and regulatory needs. Regulatory Compliance Associates works with international Fortune 100 companies, venture capital start ups, and companies of all sizes and shapes. our compliance enforcement solutions for law firms include remediation for warning letters, FDA 483’s, import bans or consent decrees. Very few regulatory compliance services have the same regulatory compliance expertise in a variety of medical fields.
Cybersecurity
For medical device manufacturers, technology can be a double-edged sword. The innovative technologies that elevate the quality of life for patients can also be used to potentially undermine the organization using the device. The consequences can affect the device itself if Regulatory Compliance Associates medtech consultants do not implement good IoT cybersecurity and FDA cybersecurity protocols.
At Regulatory Compliance Associates, we offer a wide variety of services for medical devices security to help ensure that your product is protected from cyber-attacks. With a well-planned design, along with full visibility of product development and the supply chain, Regulatory Compliance Associates medical device consultant Experts can help strengthen your device’s cybersecurity. We partner with medical device companies in each phase of the design cycle, including protecting inputs from threat exposure and hardening outputs for regulatory compliance & FDA submission approval of your medical technology.
Regulatory Affairs
Regulatory affairs is Regulatory Compliance Associates® backbone, and we handle more submissions in a month than many manufacturers do in a lifetime. Our regulatory compliance consulting Experts have experience working with the FDA, global regulatory bodies and / or agencies, and notified bodies worldwide. Therefore, you can count on us for in-depth and up-to-date insights which increase speed-to-market.
As a trusted regulatory affairs consultant, our FDA veterans and industry experts represent Regulatory Compliance Associates® as one of the top medical device consulting firms. We’re here to help you navigate the difficulties associated with new product submissions. Regulatory Compliance Associates® medical device consulting company has expertise in both the approval process and post-approval support.
- New Product Approval
- Post-Approval Support
- Outsourced Staffing
- EU MDR
- Combination Products
Compliance Assurance
Increasingly, life science companies are feeling the pressure of greater scrutiny by regulators, and responding by developing sustainable compliance strategies. Whether it’s preparing for an audit, developing a response to an FDA finding, or remediation to an adverse event, Regulatory Compliance Associates® can help.
Our network of over 500 medical device consultant & FDA, MHRA & EMA veterans are industry professionals offers a unique blend of expertise. This allows Regulatory Compliance Associates® to handle both simple and complex regulatory compliance challenges within medical device consulting companies.
- Gap Assessments
- Internal Audits
- Employee Training
- Notified Body Response
- Data Integrity
Quality Assurance
Regulatory Compliance Associates® Quality Assurance consulting includes quality system assessments, strategy, implementations, and identification of quality metrics to ensure continuous improvement, aligning with your business needs and goals. Each Regulatory Compliance Associates® medical device consultant is a quality expert with experience spanning major corporations and start-ups. We know firsthand how to achieve, maintain, and improve quality, and we excel in transferring this knowledge to your organization.
In the medical devices field, quality assurance (QA) is more than merely ensuring the quality of a finished product. You need the tools to monitor and regulate every process from the design of a new product to continued quality compliance as the device is sent to market. At Regulatory Compliance Associates®, we offer you the quality assurance services you need to monitor these processes and ensure quality compliance every step of the way.
With more than 20 years experience working with medical device consulting companies, Regulatory Compliance Associates® trusted medical device quality assurance consultant team is fully equipped to handle your unique QA needs.
- ISO13485
- 21 CFR 210
- 21 CFR 211
- Outsourced Staffing
- MDSAP
- Facility Validation
- Equipment Validation
- Quality Metrics
Remediation Support
Regulatory Compliance Associates® is widely recognized within medical device consulting companies & the life science industry for remediation support. Regulatory Compliance Associates® ability to help companies successfully resolve complex regulatory challenges have a proven track record of success. Our medical device consulting services include significant experience with the development of responses to 483 Observations, Warning Letters, Untitled Letters and Consent Decrees.
- Regulatory Action
- Regulatory Compliance
- Regulatory Enforcement
- Warning Letter
- 483 Observation
- Oversight Services
Our value goes beyond the initial response by helping companies successfully execute their action plans, develop an improved compliance culture tailored to the needs of their business, and ultimately move beyond the regulatory action to emerge as a stronger business. We negotiate difficult demands of remediation with insight and the clear advantage of our medical device consultant expertise and experience that makes partnering with Regulatory Compliance Associates® a competitive differentiator in the remediation space.
- Quality System
- Technical File
- Design History File
- Data Integrity
- cGMP
Strategic Consulting
Whether it’s a strategy, a technical plan, or project, Regulatory Compliance Associates® medical device consultancy can help ensure a successful project. Regulatory Compliance Associates® medical device strategy consulting can deliver your project on time, on budget, and you’re never embroiled in a costly mistake.
Our medical device consultant Experts are industry Experts are here to provide the unique insight you need before an M&A deal, through a staffing crisis and in every area of your product’s development and life cycle. As the trusted medical device manufacturing consultants of thousands of companies around the world, we have the knowledge and expertise needed to deliver exceptional results to your business — no matter your size or unique needs.
- Manufacturing Optimization
- Product Lifecycle Management
- Mergers & Acquisitions (M&A)
- Due Diligence
- Device Vigilance
- Risk Management Plan
- Product Complaints
- Medical Information
About Regulatory Compliance Associates
 Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
- Life Sciences
- Pharmaceutical
- Biologic & Biotechnology
- Sterile compounding
- Medical device
- Lab Testing
We understand the complexities of running a life science business and possess areas of expertise that include every facet of R&D, operations, regulatory affairs, quality, and manufacturing. We are used to working on the front lines and thriving in the scrutiny of FDA, Health Canada, MHRA and globally-regulated companies.
As your partners, we can negotiate the potential minefield of regulatory compliance and regulatory due diligence with insight, hindsight, and the clear advantage of our unique expertise and experience.
- Founded in 2000
- Headquartered in Wisconsin (USA)
- Expertise backed by over 500 industry subject matter experts
- Acquired by Sotera Health in 2021
About Sotera Health
The name Sotera Health was inspired by Soteria, the Greek goddess of safety, and reflects the Company’s unwavering commitment to its mission, Safeguarding Global Health®.
Sotera Health Company, along with its three best-in-class businesses – Sterigenics®, Nordion® and Nelson Labs®, is a leading global provider of mission-critical end-to-end sterilization solutions and lab testing and advisory services for the healthcare industry. With a combined tenure across our businesses of nearly 200 years and our industry-recognized scientific and technological expertise, we help to ensure the safety of over 190 million patients and healthcare practitioners around the world every year.
We are a trusted partner to more than 5,800 customers in over 50 countries, including 40 of the top 50 medical device companies and 8 of the top 10 pharmaceutical companies.
Commitment to Quality
Our Certificate of Registration demonstrates that our Quality Management System meets the requirements of ISO 9001:2015, an internationally recognized standard of quality.
To begin the Regulatory Compliance Associates® scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage.
Connect with RCA Today
Contact us to learn more about our regulatory compliance experts and how they can help
