















Published Articles

The main regulatory standard for ensuring pharmaceutical quality is 21 Code of Federal Regulations (CFR) Parts 210 and 211, collectively referred to as the current good manufacturing practice (CGMPs) regulation for human pharmaceuticals. It’s not only the CGMP regulations that matter; the approaches biopharmaceutical companies take to interpret and embrace these regulations are of equal, if not more, importance. The remediation costs associated with CGMP non-compliance can be staggering to say the least.
CGMPs place emphasis on product quality and compliance with the regulations. So how do companies embrace, and even embody, quality and compliance with CGMPs? One way minimize remediation costs is to operate under a quality culture.
Quality Culture
A company should have a pharmaceutical quality system as described in FDA Guidance for Industry, Q10 Pharmaceutical Quality System. A quality culture is created when managers believe a company has a duty to create a mutually beneficial relationship between itself, its employees, and its customers.
Culture is the shared beliefs, values, attitudes, and behavior patterns that characterize a family, a community, or an organization. A healthy organizational culture is rooted in the understanding that quality is good for the company and its customers. Thus, its existence is a driving force behind how employees act and behave regardless of level, title, or decision-making authority.
FDA Form 483
In defining a set of Six Sigma values for a corporate culture, the core principles should include integrity, customer focus, and people. This type of quality culture begins with company leaders who believe in the necessity of serving customers in order for their organizations to succeed. Six Sigma is a widely accepted methodology to help reduce remediation costs.
FDA conducts several types of inspections to help protect consumers from unsafe products: pre-approval inspection, routine inspections of a registered facility, and “for-cause” inspections. After FDA completes an inspection, company management may receive an FDA Form 483 when an investigator(s) has observed any conditions that, in their judgment, may constitute violations of the Food Drug and Cosmetic (FD&C) Act, related acts, and applicable sections of 21 CFR 210 and 211.
FDA Warning Letter
Observations are made when, in the FDA investigator’s judgment, conditions or practices observed would indicate that product has been adulterated or is being prepared, packed, or held under conditions whereby it may become adulterated or rendered injurious to health.
FDA Form 483 notifies the company’s management of objectionable conditions. Companies respond to the 483 in writing with their corrective action plan and implement schedule. The 483 is closed when the company receives their establishment inspection report (EIR). Unfortunately, there are companies that either do not follow through on their commitments or they do so too slowly. If circumstances merit, FDA can choose to escalate the potential for remediation costs by serving the company with a warning letter.
CAPA
Typically, FDA gives individuals and companies an opportunity to take voluntary and prompt corrective action before the agency initiates an enforcement action. The primary goal of a CAPA system is to introduce new procedure(s) that address requirements for the corrective actions that return an organization to compliance. Additionally, a FDA warning letter is the principal means of notifying regulated companies of violations and corrective actions needed.
The following factors are often used to determine whether to issue a warning letter:
- The firm’s compliance history (e.g., a history of serious violations, past remediation costs or repeated failure to prevent the recurrence of violations)
- The nature of the violation (e.g., a violation that the firm was aware of [was evident or discovered] but failed to correct
- The product risk and the impact of the violations on such risk
- The overall adequacy of the firm’s corrective action and whether the corrective action addresses the specific violations. Remediation costs also may include related violations, related products or facilities, and contains provisions for monitoring and review to ensure effectiveness and prevent recurrence
- Whether documentation of the corrective action was provided to enable the agency to undertake an informed evaluation
- Whether the timeframe for the corrective action is appropriate and whether actual progress has been made in accordance with the timeframe.
Consent Decree
When a company repeatedly violates cGMP requirements, FDA can force it, through legal channels, to make specific changes. Under this severe form of escalation by FDA, the objective is no longer a discussion about responses to 483s or warning letter observations, it’s about a forced company make-over. This process, known as consent decree, exposes all broken systems within a company.
FDA does not care about any efforts and expenses undertaken by the company to redesign and implement a robust quality management system; a common element of a consent decree is demonstrating sustainability. A company under consent decree who doesn’t demonstrate sustainability can lose future revenue in different ways. One way, which is particularly difficult to quantitate, is the price of “lost innovation”. The company is redirecting revenue into compliance, oversight, and remediation instead of reinvesting it into research and development of new products.
Consent Decree Definition
As previously mentioned, Sustainability is one of the primary drivers for defining an organization’s capacity to know when it is veering off course. The consent decree definition of the program is critical to making the correct decisions, taking appropriate actions, and maintaining a state of control without future external intervention.
Sustainability embraces the core quality culture values and expected behaviors of integrity, empowerment, and accountability. A consent decree mandates a series of annual inspections performed by a third-party to monitor sustainability. FDA recommends companies hire external experts and invest time and money to inspect and certify compliance, often for many years.
Operating under a consent decree is a dire situation for the company and one where there is no certain predictability of the outcome. To understand the full magnitude of a consent decree’s impact, one needs to take into consideration the many and various modes in which the negative consequences can be realized (see Figure 1).
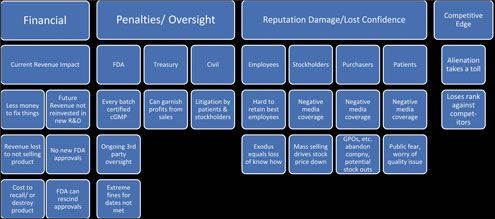
Figure 1: Overview of different repercussions
to and reverberations from a pharma company
operating under consent decree. (Image courtesy of author)
Remediation Costs of Non-Compliance
This discussion focuses on three ways to look at the costs of non-compliance:
- Quantifiable costs
- Difficult-to-quantify costs
- Invisible costs with hidden impacts
Quantifiable Costs
Quantifiable costs are those that can have a value assigned to them with a reasonable degree of accuracy. Quantifiable costs are usually more obvious, but they manifest in several ways as shown in Figure 1.
A major requisite term of a consent decree is for the company to retain, at its own expense, an independent third-party for ongoing certification and oversight of the implementation of agreed to corrective action. Another requirement may be a commitment to have every released batch certified to be CGMP.
At Will Employment
At Will Employment is defined as an indefinite period of time of employment that may be terminated either by employer or employee. It is not uncommon that firing an employee or/and replacement of multiple employees is a step associated with a consent decree. The training and retraining costs can often be difficult to quantify in the short term until compliance improves. This company action is meant to set the tone internally that the non-compliant way of doing business will no longer be tolerated.
FDA Violation Penalties
FDA can levy significant violation penalties in the form of fines for not meeting action dates. Examples include $15,000 per day for missed dates, royalty payments up to 24.6% per product not revalidated on time, and costs for FDA inspections. Furthermore, the US Treasury can garnish profit from sales through fines. Such was the case for Wyeth, who agreed to a consent decree regarding its Marietta, PA and Pearl River, NY facilities. FDA inspections of these facilities found several GMP deviations that eventually resulted in warning letters.
One of the largest drug safety settlements occurred when generic-drug manufacturer, Ranbaxy USA Inc., a subsidiary of Indian generic-pharmaceutical manufacturer Ranbaxy Laboratories Limited, pleaded guilty to felony charges relating to the manufacture and distribution of certain adulterated drugs made at two of Ranbaxy’s manufacturing facilities in India (3). Ranbaxy paid a criminal fine and forfeiture totaling $150 million and settled civil claims under the False Claims Act and related state laws for $350 million.
Civil Monetary Penalties Law
Entering into consent decree can also expose a company to civil penalties. Depending on the circumstances, shareholders, patients, and sometimes even company employees may be able to sue for damages.
One such law suit arose from an unsuccessful effort by Baxter International to fix problems with its Colleague Infusion Pump. Westmoreland County Employee Retirement System (Westmoreland) alleged that Baxter’s directors and officers breached their fiduciary duties by “consciously disregarding their responsibility to bring Baxter into compliance with the 2006 consent decree and related health and safety laws”.
FDA Recall
The Baxter breach was alleged to have caused the organization to lose more than $550 million after FDA mandated a recall of the Colleague Infusion Pumps. Baxter invested time and money trying to fix the pumps, but the manufacturing problems persisted over time.
FDA invoked its power under the original consent decree by ordering Baxter to recall and destroy all Colleague Infusion Pumps then in use in the US. This is where invisible costs with hidden impacts turn into the customer reimbursement for the value of the recalled device. Invisible costs also included Baxter being required to assist in finding replacement devices for those customers impacted. The company’s stock price fell by more than 4% after the announcement and the company later recorded a pre-tax charge of $588 million to account for the estimated costs of the recall.
Drug Shortage
When a drug manufacturer subject to enforcement action is the sole supplier of an important medicine, drug shortages become a concern. Short-term solutions may include doctors substituting medications that may have lesser efficacy. Pharmaceutical companies with potential manufacturing capability may be incentivized by FDA to manufacture identical or equivalent drug products; however, medium-to-long lead times can delay product availability. When supplies dwindle, patients must pay higher prices for the same drug.
One illustration of this scenario is the FDA consent decree with Genzyme regarding repeated manufacturing issues at the company’s Allston, MA facility. The consent decree included an up-front disgorgement of past profits of $175 million and the requirement to move fill/finish operations out of the Allston plant by a specific date. Had Genzyme not met those deadlines, FDA could have required the company to disgorge 18.5% of revenue for the affected products.
Difficult-to-Quantify Costs
Other financial ramifications from consent decrees may be difficult to quantify. For example, some employees may lose their confidence in the commitment or ability of the company’s CEO and other executives to manage the situation.
Employee Attrition
Employee attrition is expected, but if the exodus includes long-tenured employees the company may be drained of valuable knowledge and talent. The longer the situation exists, the more difficult it becomes to retain the best employees.
Reputation Damage
A company under a consent decree is subject to reputation damage, which can be accentuated by the ease of publicly available negative media coverage about how the company operated. Negative media coverage can result in public fear. In the Ranbaxy case, an import ban had been in place for 30 drugs manufactured at two of its Indian manufacturing plants resulting from alleged data falsification. This type of information can result in public concerns that medicines could be adulterated.
Batch Release
Lost revenue due to the company’s inability to sell product can also be an issue. Companies under consent decree experience long delays in releasing product due to the intensive oversight required, when a batch fails release testing, and when a product is recalled. The cost for the logistics of product recall and destruction can be high, especially if the API is expensive or has a long lead time.
Group Purchasing Organizations
Another potential cost is the inability to sell a product. Group purchasing organizations (GPOs) use the power of collective purchasing to buy pharmaceuticals at discounts. An underlying premise is continuity of supply; if a pharmaceutical manufacturer is unable to deliver the product contracted, the GPO must obtain replacement product from the open market, often at a higher price. GPOs wary of a potential inability of the company to supply its products may consider other sources.
In one of the largest settlements to date, Ranbaxy pled guilty to felony charges. The charges were manufacturing and distributing adulterated drugs made at the Indian manufacturing sites. The criminal fine and forfeiture totaled $150 million and another $350 million in remediation costs to settle civil claims under the False Claims Act.
Invisible Costs and Hidden Impacts
If the remediation costs to bring a facility under consent decree into CGMP compliance is determined to exceed the company’s financial ability or it makes no financial sense to continue operations, the company’s management may decide to close the facility. Shuttering a facility, can have devastating impact on the local community, particularly in rural areas where the manufacturer is a primary employer. The community’s tax base will also suffer.
The company can lose its competitive edge because its busy focusing inside rather than externally. This can be a competitive advantage to your closest competition looking for a way in which to leverage your situation for their benefit. The potential opens for the company to become alienated and therefore lose rank compared to its direct competitors.
A company can also be denied approval of new drug while non-compliance exists (4). Because of its consent decree, approval of two new drugs by Eli Lilly was delayed. This was the result of CGMP issues being found during pre-approval inspection in the fall of 2001, which was six years after its consent decree.
Conclusion
The collective cost of remediation of non-compliance far exceeds the remediation cost to remain in compliance. The number of consent decrees issued per year has remained consistent during the past decade. However, companies have found it difficult to extricate themselves from the agreements.
As a result, the number of companies under consent decrees at any given time has increased. Generally, it takes many years for a company to demonstrate that it is in full compliance. Only one company that has received a decree in the past 10 years has met all requirements and had the decree lifted.
Considering the fines and the payments to the third-party consultants, the remediation costs associated with a consent decree can become very high and have a significant effect on a company’s profit. It is estimated that the costs incurred by Warner-Lambert for a consent decree–in terms of product terminations, delays in approvals, and bringing facilities and systems into compliance–was nearly $1 billion. The Warner Lambert fine was only $10 million, a small percentage of the total cost. Schering-Plough’s initial fine was $500 million. Abbott Laboratories has spent almost $1 billion resulting from a consent decree issued, including a fine of approximately $100 million.
About the Article
![]()
Pharmaceutical Technology
Vol. 41, No. 11
Pages: 54–57
To begin the Regulatory Compliance Associates scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage. You may also email us at [email protected].
Connect with RCA Today
Contact us to learn more about our regulatory compliance experts and how they can help
