

















Blog
The U.S. Government Accountability Office (GAO) publishes independent audits at the request of consumer advocates, watchdog groups, and members of the United States Congress. One of the most recent publications involved the U.S. Food and Drug Administration (FDA) post market surveillance process, including an analysis of management & oversight across agency functions and employees.
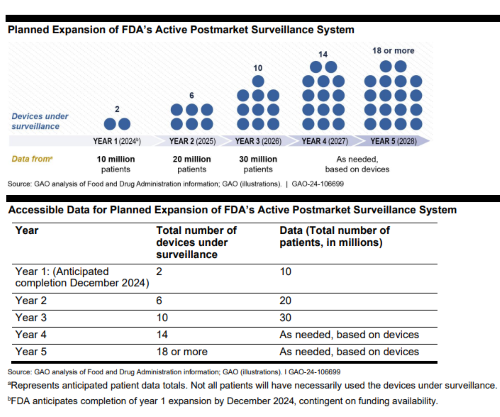
Additionally, the post marketing study uncovered hurdles FDA encountered during the medical device audit. The final 27-page report included feedback on whether the agency was efficient while handling problems and protecting data integrity. These details offer a snapshot of how a regulatory body operates in real time across the life science industry, and where efficiency could be examined.
What is a Post Market Surveillance System?
The U.S. Food and Drug Administration (FDA) has an approved regulatory framework & database (MAUDE) designed to connect consumer complaints and compliance adverse events to a product code and medical device manufacturer. This post market surveillance system (PMS) is a validated FDA regulatory process designed to increase transparency and continuous improvement.
Adverse Events
A post market surveillance system is the market listening mechanism between real world evidence and analysis. Software platforms are used as a global harmonization resource for monitoring adverse events (e.g. poisoning, adverse reaction) across countries, devices & the cycle of patient life.
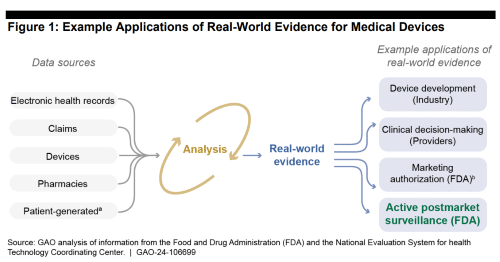
Additionally, the post market surveillance system is designed to help collect HIPAA-compliant patient data & medical device product ID info in a single source of postmarket truth. Patent-generated evidence (e.g. FitBit, Apple Watch) is a growing data source HCPs recognize at the point of care which medical device manufacturers could use during product design & commercialization.
Patient Safety
Additionally, the post marketing surveillance process uses incoming data to search for patient safety trends proactively. The future may include post marketing requirements that help educate the population’s health, and when label changes should be communicated to consumers. Finally, when a negative safety trend begins to accelerate, FDA may consider if the marketing approval of the product should be removed.
What is a Unique Device Identifier (UDI)?
The unique device identification (UDI) is an exclusive code related to a specific medical device. Each UDI promotes a transparent identification system that helps life science employees trace a product using a numerical & alphabetical format on the device label. Each unique device identification number includes a mix of production and device identifier (DI) data based on the manufacturing process. Further, the elements of the production identifier (PI) portion of the UDI code include this information on each device label:
- Manufacturing date
- Manufacturing batch number (or lot number)
- Device serial number
- Device expiration date
- Distinct identification code (for combination products)
FDA mandates that any patient documentation needed to uncover the root cause of a medical device’s problem be included in the safety profile risk analysis. Ideally, the risk analysis would also contain information from a patient’s EHR health records and/or the payer’s claim data or pharmacy data. Both of these are widely considered reliable post market surveillance data sources.
A full transcript and highlights of the GAO report can be found here.
