

















Blog
Why Design Controls Are the Biggest Leap for Drug Teams
For pharmaceutical and biologic organizations entering the combination product space, few regulatory requirements cause as much friction as design controls under FDA QMSR. Unlike traditional pharmaceutical Quality Risk Management (QRM), design controls require a structured and documented history of how the device portion of a product was conceived, designed, verified, validated, and transferred into production.
For many drug teams, this represents a fundamental shift from the line of thinking they are accustomed to. Quality is no longer focused solely on batch release, clinical outcomes, or GMP compliance. Instead, regulators expect a living design narrative that demonstrates intentional engineering decisions and risk-based controls throughout the product lifecycle.
Why Pharmaceutical Risk Management Is Not Device Risk Management
Pharmaceutical QRM, often aligned with ICH Q9, is typically focused on patient safety, process variability, and manufacturing controls. Device risk management, however, is governed by ISO 14971 and embedded directly into design controls.
The key distinction is timing and intent.
Pharma risk management often evaluates risk after formulation and process decisions are made. Device risk management is proactive and iterative, requiring teams to identify hazards, estimate and evaluate risks, implement controls, and reassess residual risk throughout design and development.
For combination products, the FDA expects risk management to be tightly linked to design inputs, outputs, verification, and validation. A standalone risk assessment or FMEA is not sufficient if it is not clearly integrated into the design history.
The Circular Nature of Design Inputs and Outputs
One of the most misunderstood aspects of design controls is that design is not linear. Design inputs inform outputs, but outputs frequently expose gaps, ambiguities, or risks that require refinement of inputs. This circular relationship is not a weakness. It is an expected and documented part of compliant device development.
Design inputs define what the device must do, including performance requirements, safety considerations, usability needs, and regulatory constraints. Design outputs translate those requirements into drawings, specifications, software code, and manufacturing instructions.
FDA investigators routinely look for evidence that inputs and outputs were revisited as new information emerged. Static inputs that never evolve raise red flags during inspection.
Why Clinical Trials Do Not Equal Design Validation
A common misconception among drug-focused organizations is that clinical trials satisfy device design validation requirements. While clinical data may support aspects of performance or safety, FDA does not consider clinical trials alone to be sufficient design validation for the device component.
Design validation under QMSR and ISO 13485 requires documented evidence that the device meets user needs and intended uses under actual or simulated use conditions. This includes usability testing, human factors engineering, and validation of device performance independent of clinical efficacy endpoints.
In short, proving the therapy works does not automatically prove the device was designed correctly.
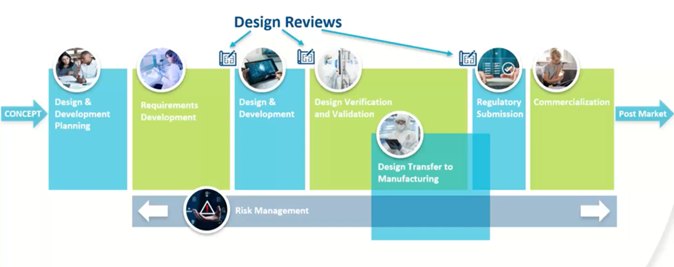
Design Transfer Is Where Many Programs Fail
Design transfer is often treated as an administrative milestone. In reality, it is a critical control point where FDA scrutiny increases significantly.
Design transfer ensures that the device design is correctly translated into production specifications and manufacturing processes. Regulators expect clear evidence that manufacturing can consistently produce devices that meet approved design requirements.
Weak design transfer frequently results in downstream CAPAs, nonconformances, and delayed approvals. Strong design transfer, on the other hand, creates alignment between R&D, quality, and manufacturing and significantly reduces lifecycle risk.
Building Design Controls Without Rebuilding Your QMS
For combination products, design controls do not require a full medical device QMS. Under the streamlined approach, organizations can integrate design control elements into an existing pharmaceutical system while maintaining compliance with 21 CFR Part 4.
The key is intentional integration, not duplication. Design controls must be documented, traceable, and inspection-ready, but they can coexist with pharmaceutical processes when structured correctly.
How RCA Helps Drug Teams Navigate Design Controls
Design controls are challenging because they force organizations to think differently about development. Regulatory Compliance Associates® (RCA) helps pharmaceutical and biologic companies implement right-sized design control frameworks that meet FDA expectations without unnecessary complexity.
Ready to streamline your QMS for combination product success?
Contact RCA today to schedule a consultation and take the first step toward regulatory readiness and market leadership.
