
















Click to watch Regulatory Compliance Associates medical device consultant, Dr. Stephen Coulter, discuss the common tools & processes used during product development. Early stages of new product development include analyzing user needs & translating design benefits into long-term patient efficacy.
Developing an original product idea is difficult but many common tools & processes are consistently used. The early stages of product development include analyzing user needs and how those translate into design inputs.
Product Development Strategy
Ideation and product development life cycle research can help you identify user needs. Additionally, increasing the quality of an existing product is often a successful new product development strategy. Your solution doesn’t have to be a radically new idea.
Does it have a substitute product that can take market share? Or, can you combine the features of multiple products during the product design process to make a combination product that patients prefer more? Could a product design consultant help answer these questions & provide knowledge transfer?
Product Development Cycle
Start by asking your cross-functional team an initial question: “What do we need to change to achieve success during the product development cycle?”. Product development strategy includes modifying existing systems & processes to accelerate efficiency. Refine your model or process based on the unique types of product solutions in your pipeline.
Product Research
Being able to understand & explain why your solution addresses a user needs statement is inherent to launch sales success. Interactive activities can be useful for improving creative conversation during market development strategy sessions. Further, voice of the customer (VOC) research consistently helps target the patient population in mind.
Finally, using VOC to identify early adopter audiences & the audience’s most important needs can increase early interest in your solution. Begin the sales process by determining core messaging ahead of the launch ramp-up based on device claims & if the regulatory submission is approved.
Market Development Strategy
User needs are the foundation of any successful market development strategy. Yet, being able to commercialize a new solution based on user needs using a team experiencing process problems is never an easy task. It requires the Engineering Team & Operations Team at the listening table earlier in the process than later.
Product Design and Development
A critical to quality objective early in the new product development process is removing everything but the essentials. Research your users needs to create design inputs for a product that patients both need & generates revenue.
Product Planning Process
After a business case is determined in the waterfall method, the product planning process begins cross functionally to validate product ideas via new product development research. What user needs will accelerate a successful product launch the fastest?
Innovative product design means addressing honest opinions from your target audience to validate your medical device solves a problem. The product development management team must be convinced the product can win in the commercial marketplace sooner rather than later.
Software Product Design
Success during the launch of a software as a medical device is often found at the heart of the product development plan. Any new product design must incorporate value added for the audience in one way, shape or form. This can include lean product design as some of the most effective software in the industry is not complicated.
Software as a Medical Device
Consider the regulatory differences between a tangible product and a technology product very carefully. For example, Software as a Medical Device (SaMD) has changed the marketplace by increasing both clinical research success & primary care delivery. Include process improvement sessions during internal meetings where a collective group deciphers what the user need statements mean to their team.
Agile Product Development
The vision for development begins with a minimum viable product, or MVP, during early software conceptualization & development; what is the business case of commercialization? How do we learn faster while making real time efficiency changes?
Digital Product Development
Product development teams must answer several questions before undertaking product design. Further, one focal ask is are successful processes already in place for medical product development.
Do you have a RACI chart to show who is responsible, accountable, considered, and informed? Finally, a clear cross-functional understanding of timelines & milestones can help guide expectations during MVP product development, which can be stressful based on the speed of common work.
MVP Agile
MVP (Minimum Viable Product) a widely accepted concept in software development in digital product development. However, our medical product design discussion will take a different direction – what is the regulatory affairs process?
A project management software development team should deliver a safe, efficacious solution with the objective of delivering patient value. Define your value through a regulatory strategy based on device claims, clinical evidence, and how both help the patient population.
Regulatory Compliance
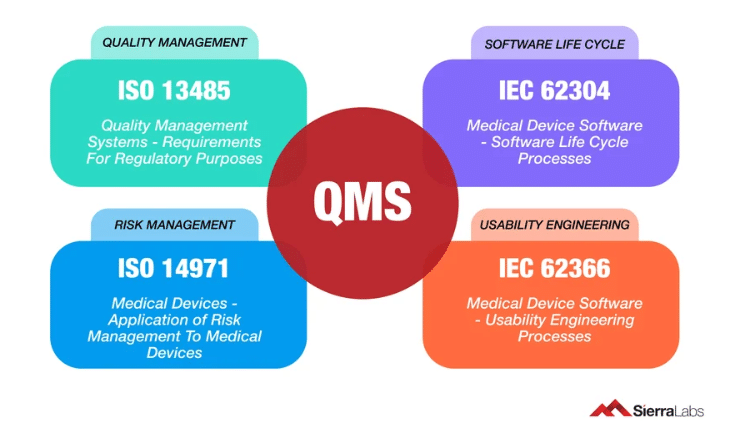
Software as a medical device involves regulatory compliance based on multiple, widely accepted global standards.
- This includes:
- ISO 13485
- IEC 62304
- IEC 62366
- ISO 14791
The product development engineering team is responsible for the software process. Any prototype development assistance should begin with a regulatory compliance perspective of claims and predicate devices.
Prototype predicate concepts begin with identifying the current risk class & why use cases help validate your proposed risk class during regulatory submission. Finally, consider younger product developers who may have never been through regulatory approval in their career. FDA readiness training can be a powerful tool that increases marketing approval & how to carefully identify and isolate risk hazards.
Regulatory Pathway
Planning the build for a product requires a regulatory pathway that includes risk management considerations based on the risk class. Create as much detail as possible during this prototyping and design inputs phase of NPD.
Product development companies, like Regulatory Compliance Associates, can help identify the correct predicate device based on a risk gap analysis. Diagrams should include material ingredients that need to be sourced to create the product. This can help you determine the risk category of the product (e.g. patient daily use, HCP specialty use, etc.) and corresponding patient hazards.


