
The International Medical Device Regulation Forum (IMDRF) recently published updated cybersecurity guidance for the medical device industry. The medical device cybersecurity working groups at IMDRF have been busy lately, publishing multiple final documents about medical devices & software as medical device (SaMD).
Regulatory Compliance
IMDRF’s medical device guidance provides steering assumptions for both regulatory compliance & medical device cybersecurity, which are appropriate for sponsors developing medical devices. Further, a primary objective of the guidance is simultaneously increasing patient safety & reducing external threats for providers and HCPs.
Global Harmonization
The guidance begins with harmonization concepts that could affect multiple departments inside a medical device manufacturer. Additionally, key areas for harmonization programs highlighted by the cybersecurity guidance include:
- Product design
- Risk management activities
- Device labelling
- Regulatory submission
- Information sharing
- Post-market activities
Product Life Cycle (PLC)
IMDRF’s cybersecurity guidance continues on with a deeper evaluation of risks associated across the product life cycle. It is recommended for potential vulnerabilities to be considered for any product life cycle stage, especially considering legacy devices that may be vulnerable to strategic risk.
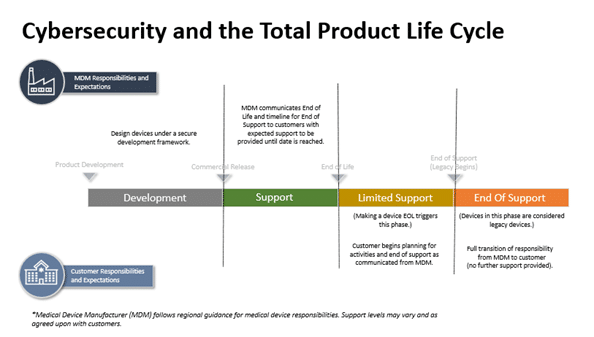
Product Design
Product design considerations include the initial phases of medical device development and continues until the end of support (EOS) once a product is discontinued. The four product design stages the cybersecurity guidance refers to when it comes to total product life cycle:
- Development Stage
- Support Stage
- Limited Stage
- End of Support
Development Stage (Stage 1)
The Development Stage occurs during the pre-commercialization phase before a medical device is approved by a regulatory body. This is when medical device manufacturers begin to incorporate security into the product concepts being designed. Design controls are critical in this stage for medical device manufacturers to leverage when considering how to mitigate risks.
Finally, an important deliverable of the Development Stage is product-related security documentation. The documentation is designed to help unfamiliar users to understand how to securely operate the medical device.
Support Stage (Stage 2)
The Support stage is during the initial post-launch phase and may continue for many years. Medical devices in this stage are:
- Currently used for providing patient care
- Available for purchase on the open market
- Contain major software, firmware, or programmable hardware components
- Support for software, firmware or components is provided by the medical device manufacturer
Additionally, medical devices in the Support stage should receive full cybersecurity support. This support often includes software patches, software updates, hardware updates, and incremental support the manufacturer considers appropriate.
Limited Support Stage (Stage 3)
Medical device manufacturers continue to provide cybersecurity support during Stage 3. However, as product development transitions to a more current medical device design, different constraints are involved with the transition. Medical devices in Stage 3 often require additional network controls compared to medical devices in Stage 2:
- Third-party components or software may be used more frequently than internally developed updates or patches
- Cybersecurity best practices integration is often governed by the ease of following support practices outlined in the Stage 2
- Medical device manufacturers must explain to users the existing limitations that are now recognized in the devices and services affected
- Healthcare providers using the medical device should begin to take more of an active role in unmitigated features of security defense.
End of Support Stage (Stage 4)
Medical devices in Stage 4 are considered more vulnerable than any of the other stages. They may still be in use for providing patient care, but they have been publicly identified as no longer being supported by the medical device manufacturer. Each of these scenarios result in a medical device that cannot be consistently defended against modern cybersecurity dangers.
Critical facets healthcare information technology departments should look for include:
- Medical devices that have been declared EOS by the medical device manufacturer
- Medical devices that are not actively marketed or sold by the medical device manufacturer
- Medical devices that contain software, firmware, or programmable hardware components no longer supported by software developers
- Medical devices with known risks to device safety and effectiveness that are unmitigated
Risk Management
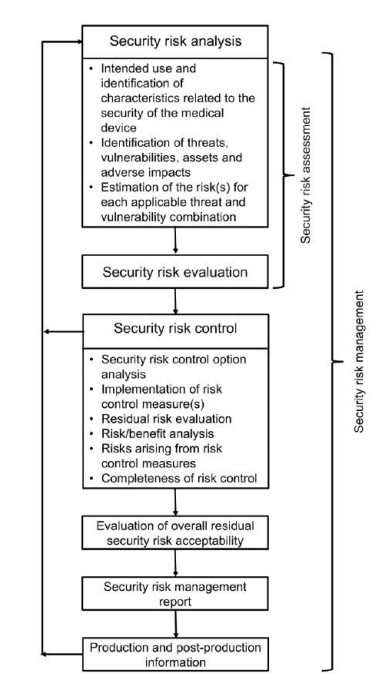 Further, the guidance calls for a risk management approach to product lifecycle management featuring:
Further, the guidance calls for a risk management approach to product lifecycle management featuring:
- Security risk analysis
- Security risk evaluation
- Security risk control
- Security risk acceptability
The cybersecurity guidance expands on product design and how security is incorporated and maintained through the product life cycle. This can be accomplished through using risk control and a secure development framework.
Risk mitigation recommendations for medical device manufacturers include:
- Security design and controls based on intended use of the medical device
- Security risk assessments across the risk management process
- Threat modelling to help determine operational risk
Security testing and communication for medical device manufacturers include:
- Customer facing product security documentation & communication
- Post-market monitoring of cybersecurity vulnerabilities
- Identification of vulnerabilities in third party risk management
- Vulnerability risk identification based on the device security design, controls, and mitigations
Ensuring availability of security patches & mitigations based on device risk:
- Coordinated and clear communication to all affected users
- Description related to the vulnerability and its corresponding mitigations
- Identification of other mitigation options when a security patch is unavailable
Data Integrity
One of the core principles the guidance stresses is cybersecurity information, data integrity and the importance of information sharing. IMDRF encourages medical device industry stakeholders to implement a proactive pre- and post-market approach to cybersecurity information sharing.
Moreover, timely information can help the industry recognize threats, evaluate associated risks, and react quickly as needed. An increase in industry transparency could directly benefit healthcare providers, medical device users and medical device companies.
Security Updates
An important section of the medical device cybersecurity guidance details stakeholder responsibilities related communications, risk management, and transfer of responsibility. Specifically, it is important that medical device manufacturer communications are comprehensive & identify types of documentation needed and when the medical device user may need it.
Product Security Documentation
Medical device manufacturers should ideally provide PLC documentation about security or support changes early in the Support stage. This helps HCP risk management during both the procurement & deployment of medical devices. Types of life cycle support for product security documentation includes:
- Manufacturer disclosure statement for medical device security
- Software Bill of Materials (SBOM)
- Security test report summaries
- Third-party security certifications
- Customer security documentation
Product Life Cycle Documentation
Medical device companies should communicate the strategic life cycle milestones to their customers. Further, these interactions would include cybersecurity EOL and EOS dates if available. This helps to support HCPs during both the procurement & installation process.
Additionally, medical device manufacturers should provide this information as far in advance as possible. The goal is at least 2 years in advance to best support healthcare professionals with the following information:
- Affected medical devices
- Medical device operating system(s)
- Version of medical device deployed
- Medical device software components
- Expected date of medical device service changes
- Extent of medical device maintenance after a service change occurs
- Additional design controls that help all involves parties
Vulnerability & Patching Information
If a vulnerability is uncovered, medical device companies should provide related vulnerability information. Further, the guidance specifically mentions the importance of both the appropriate mitigation or available software patch. Additionally, the guidance stresses an elevated priority be placed on high-risk vulnerabilities where timely communication is required. This communication is designed to help prevent both patient injury or device interruption.
Finally, the mitigation method and implementation instructions should be provided to the medical device operators. These security updates include both an over-air update or deployment of service personnel to help install the remedy.
Proactive Communications for Third-Party Components
Medical device software and other digital components within a medical device will reach EOL/EOS before the product itself does. In these cases, risk can increase based on the lack of support for these elements. To help compensate for these security risks, the cybersecurity guidance suggests medical device companies should:
- Validate the list of third-party components used in medical devices
- Track support status updates of third-party components used within their device
- Assess the risks that exist when third-party components become unsupported
- Communicate new risks and available risk mitigations to healthcare providers
About RCA’s Medical Device Consulting Services
The regulatory compliance process surrounding the medical device industry involves a strict adherence to pre/post market information throughout a device’s life-cycle. Even a single compliance issue you have can turn into a significant effect on your business. Regulatory Compliance Associates medical device consultants can help guide you through any stage of this strategic process, with capabilities during product development through the regulatory clearance/approval of your product.
Our team of over 500 medical device consulting Experts — including former FDA officials and regulatory compliance leaders in the field of medical device regulation — will work with your company to create a quality assurance and regulatory compliance approach tailored to your products and regulatory needs. Regulatory Compliance Associates works with international Fortune 100 companies, venture capital start ups, and companies of all sizes and shapes. our compliance enforcement solutions for law firms include remediation for warning letters, FDA 483’s, import bans or consent decrees. Very few regulatory compliance services have the same regulatory compliance expertise in a variety of medical fields.
Cybersecurity
For medical device manufacturers, technology can be a double-edged sword. The innovative technologies that elevate the quality of life for patients can also be used to potentially undermine the organization using the device. The consequences can affect the device itself if Regulatory Compliance Associates medtech consultants do not implement good IoT cybersecurity and FDA cybersecurity protocols.
At Regulatory Compliance Associates, we offer a wide variety of services for medical devices security to help ensure that your product is protected from cyber-attacks. With a well-planned design, along with full visibility of product development and the supply chain, Regulatory Compliance Associates medical device consultant Experts can help strengthen your device’s cybersecurity. We partner with medical device companies in each phase of the design cycle, including protecting inputs from threat exposure and hardening outputs for regulatory compliance & FDA submission approval of your medical technology.
Regulatory Affairs
Regulatory affairs is Regulatory Compliance Associates® backbone, and we handle more submissions in a month than many manufacturers do in a lifetime. Our regulatory compliance consulting Experts have experience working with the FDA, global regulatory bodies and / or agencies, and notified bodies worldwide. Therefore, you can count on us for in-depth and up-to-date insights which increase speed-to-market.
As a trusted regulatory affairs consultant, our FDA veterans and industry experts represent Regulatory Compliance Associates® as one of the top medical device consulting firms. We’re here to help you navigate the difficulties associated with new product submissions. Regulatory Compliance Associates® medical device consulting company has expertise in both the approval process and post-approval support.
- New Product Approval
- Post-Approval Support
- Outsourced Staffing
- EU MDR
- Combination Products
Compliance Assurance
Increasingly, life science companies are feeling the pressure of greater scrutiny by regulators, and responding by developing sustainable compliance strategies. Whether it’s preparing for an audit, developing a response to an FDA finding, or remediation to an adverse event, Regulatory Compliance Associates® can help.
Our network of over 500 medical device consultant & FDA, MHRA & EMA veterans are industry professionals offers a unique blend of expertise. This allows Regulatory Compliance Associates® to handle both simple and complex regulatory compliance challenges within medical device consulting companies.
- Gap Assessments
- Internal Audits
- Employee Training
- Notified Body Response
- Data Integrity
Quality Assurance
Regulatory Compliance Associates® Quality Assurance consulting includes quality system assessments, strategy, implementations, and identification of quality metrics to ensure continuous improvement, aligning with your business needs and goals. Each Regulatory Compliance Associates® medical device consultant is a quality expert with experience spanning major corporations and start-ups. We know firsthand how to achieve, maintain, and improve quality, and we excel in transferring this knowledge to your organization.
In the medical devices field, quality assurance (QA) is more than merely ensuring the quality of a finished product. You need the tools to monitor and regulate every process from the design of a new product to continued quality compliance as the device is sent to market. At Regulatory Compliance Associates®, we offer you the quality assurance services you need to monitor these processes and ensure quality compliance every step of the way.
With more than 20 years experience working with medical device consulting companies, Regulatory Compliance Associates® trusted medical device quality assurance consultant team is fully equipped to handle your unique QA needs.
- ISO13485
- 21 CFR 210
- 21 CFR 211
- Outsourced Staffing
- MDSAP
- Facility Validation
- Equipment Validation
- Quality Metrics
Remediation Services
Regulatory Compliance Associates® is widely recognized within medical device consulting companies & the life science industry for remediation services & support. Regulatory Compliance Associates® ability to help companies successfully resolve complex regulatory challenges have a proven track record of success. Our medical device consulting services include significant experience with the development of responses to 483 Observations, Warning Letters, Untitled Letters and Consent Decrees.
- Regulatory Action
- Regulatory Compliance
- Regulatory Enforcement
- Warning Letter
- 483 Observation
- Oversight Services
Our value goes beyond the initial response by helping companies successfully execute their action plans, develop an improved compliance culture tailored to the needs of their business, and ultimately move beyond the regulatory action to emerge as a stronger business. We negotiate difficult demands of remediation with insight and the clear advantage of our medical device consultant expertise and experience that makes partnering with Regulatory Compliance Associates® a competitive differentiator in the remediation space.
- Quality System
- Technical File
- Design History File
- Data Integrity
- cGMP
Strategic Consulting
Whether it’s a strategy, a technical plan, or project, Regulatory Compliance Associates® medical device consultancy can help ensure a successful project. Regulatory Compliance Associates® medical device strategy consulting can deliver your project on time, on budget, and you’re never embroiled in a costly mistake.
Our medical device consultant Experts are industry Experts are here to provide the unique insight you need before an M&A deal, through a staffing crisis and in every area of your product’s development and life cycle. As the trusted medical device manufacturing consultants of thousands of companies around the world, we have the knowledge and expertise needed to deliver exceptional results to your business — no matter your size or unique needs.
- Manufacturing Optimization
- Product Lifecycle Management
- Mergers & Acquisitions (M&A)
- Due Diligence
- Device Vigilance
- Risk Management Plan
- Product Complaints
- Medical Information
About Regulatory Compliance Associates
 Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
Regulatory Compliance Associates® (RCA) provides medical device consulting to the following industries for resolution of life science challenges:
- Life Sciences
- Pharmaceutical
- Biologic & Biotechnology
- Sterile compounding
- Medical device
- Lab Testing
We understand the complexities of running a life science business and possess areas of expertise that include every facet of R&D, operations, regulatory affairs, quality, and manufacturing. We are used to working on the front lines and thriving in the scrutiny of FDA, Health Canada, MHRA and globally-regulated companies.
As your partners, we can negotiate the potential minefield of regulatory compliance and regulatory due diligence with insight, hindsight, and the clear advantage of our unique expertise and experience.
- Founded in 2000
- Headquartered in Wisconsin (USA)
- Expertise backed by over 500 industry subject matter experts
- Acquired by Sotera Health in 2021
About Sotera Health
The name Sotera Health was inspired by Soteria, the Greek goddess of safety, and reflects the Company’s unwavering commitment to its mission, Safeguarding Global Health®.
Sotera Health Company, along with its three best-in-class businesses – Sterigenics®, Nordion® and Nelson Labs®, is a leading global provider of mission-critical end-to-end sterilization solutions and lab testing and advisory services for the healthcare industry. With a combined tenure across our businesses of nearly 200 years and our industry-recognized scientific and technological expertise, we help to ensure the safety of over 190 million patients and healthcare practitioners around the world every year.
We are a trusted partner to more than 5,800 customers in over 50 countries, including 40 of the top 50 medical device companies and 8 of the top 10 pharmaceutical companies.
Commitment to Quality
Our Certificate of Registration demonstrates that our Quality Management System meets the requirements of ISO 9001:2015, an internationally recognized standard of quality.
To begin the Regulatory Compliance Associates® scoping process today, please enter your information in the blue form below and click the submit button at the bottom of the webpage.
 The FDA harmonization team has recently announced it is no longer going to be a member of the Global Harmonization Working Party (GHWP). This comes as an eye-opener for many life science industry employees since FDA harmonization has been a visible priority since the agency joined GHWP in 2021.
The FDA harmonization team has recently announced it is no longer going to be a member of the Global Harmonization Working Party (GHWP). This comes as an eye-opener for many life science industry employees since FDA harmonization has been a visible priority since the agency joined GHWP in 2021. Regulatory Compliance Associates® (
Regulatory Compliance Associates® (